 Revista Fisiopatología y Diagnóstico Médico Revista Digital bimestral
Revista Fisiopatología y Diagnóstico Médico Revista Digital bimestral
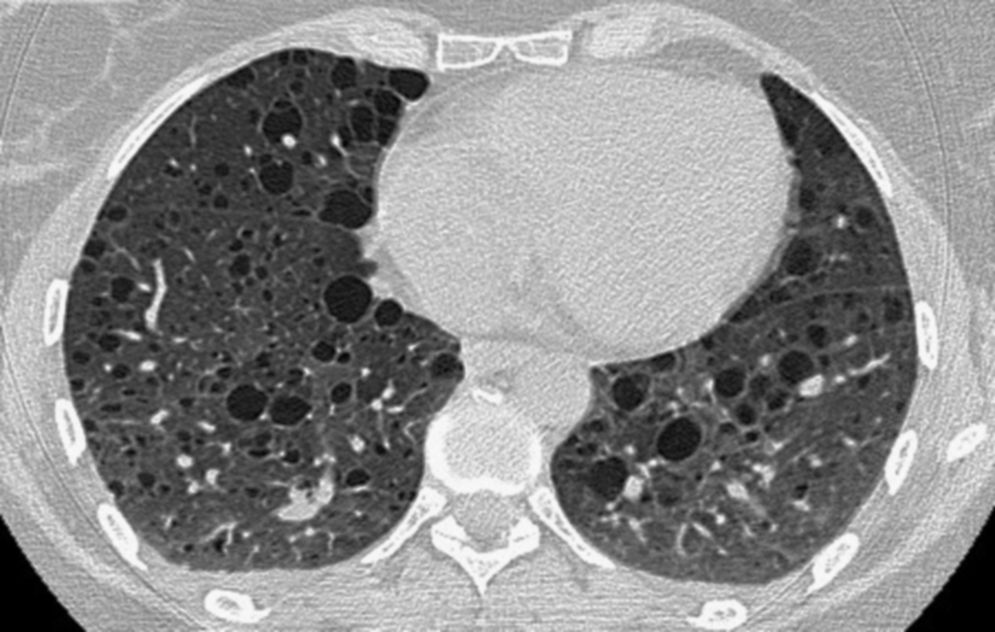
Articulos Relacionados



ES 1 jefferson intriago 04 06 2017
Introducción
Una enfermedad rara se define como aquella que afecta al menos 5 por cada 10.000 habitantes en el mundo. Las enfermedades huérfanas son aquellas para las que no existe una investigación amplia, no se dispone de tratamientos específicos, y solo tienen un interés limitado para científicos y médicos. En consecuencia, los pacientes se sienten abandonados y «huérfanos» en el mundo de la asistencia sanitaria. Las enfermedades huérfanas pueden ser comunes o raras. (Europan Lung Fundation, 2013) La Linfangioleiomiomatosis pulmonar fue notificada por primera vez en 1918 por el Dr. Lutembacher en una paciente con esclerosis tuberosa o complejo esclerosis tuberosa (CET o TSC). El CET es una enfermedad genética autosómica dominante que se presenta en 1 de cada 6000 nacimientos, se caracteriza por la formación de tumores benignos en múltiples órganos, y en la actualidad afecta a más de un millón de personas en todo el mundo. Es importante tener en cuenta que cerca del 40% de mujeres con CET pueden padecer LAM. Fue en 1937 cuando Von Stossel informó de la existencia de linfangioleiomiomatosis esporádica (LAM), es decir que no estaba asociada a una enfermedad genética subyacente. Gracias a Sue Byrnes, madre de una paciente diagnosticada de LAM, que en 1995 decidió hacer lo imposible para encontrar una cura a la enfermedad de su hija, creando la primera y mayor organización de LAM en el mundo: The LAM Foundation. En este tiempo, ha recaudado millones de dólares destinados a investigación con los que se ha logrado identificar un gen LAM, conocer el porqué del crecimiento anormal de células de músculo liso en LAM, y el primer ensayo de un medicamento para la linfangioleiomiomatosis, el MILES TRIAL.(Meritxell Lupon, 2012)

