 Revista Fisiopatología y Diagnóstico Médico Revista Digital bimestral
Revista Fisiopatología y Diagnóstico Médico Revista Digital bimestral
Articulos Relacionados



ES6 andrea palacios 28 08 2017
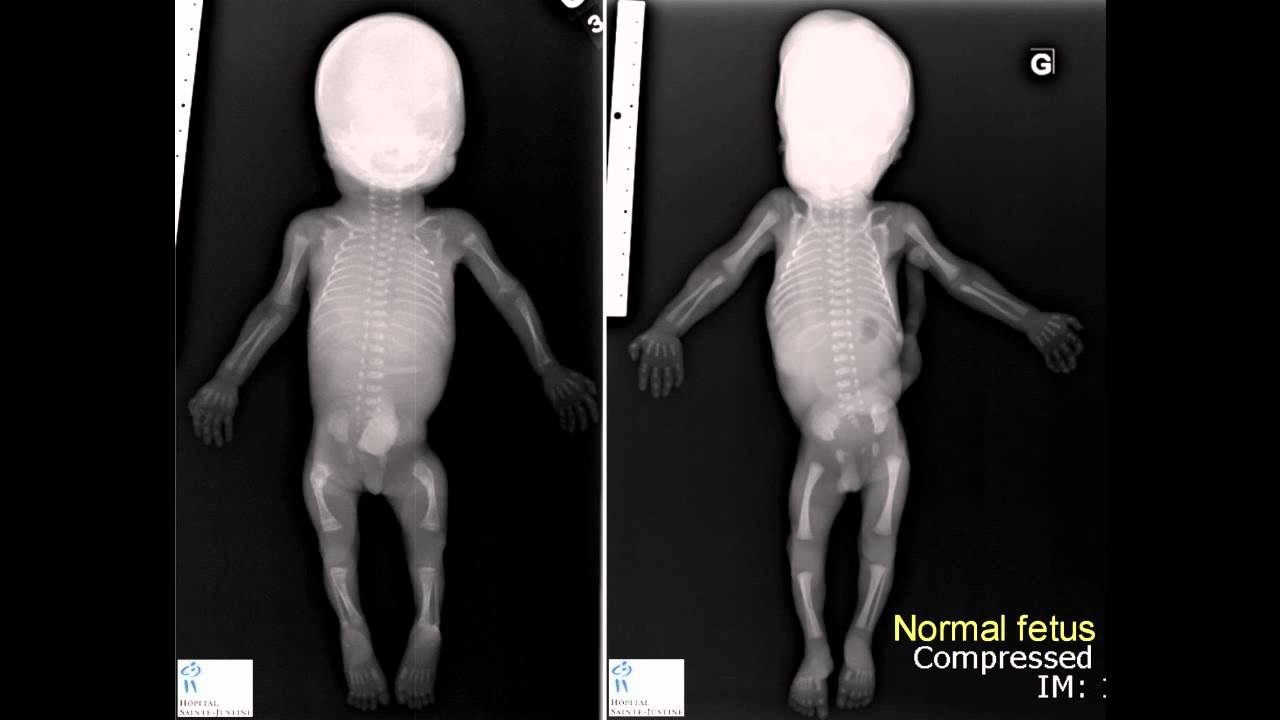
La Osteogénesis Imperfecta (OI) es una entidad de origen genético que causa un defecto en la formación de colágeno tipo I, caracterizada por la fragilidad ósea y con un espectro clínico variable desde un escaso número de fracturas hasta deformidades esqueléticas severas con resultados fatales. (Lazala y Solaque 2009) El colágeno tipo I es una proteína presente en todos los tejidos de sostén, especialmente en el hueso (donde el colágeno tipo I es el principal componente de la matriz orgánica), piel y tendones, pero también en ligamentos, fascias, córnea, esclera, dentina y vasos sanguíneos. Las consecuencias de la alteración de este colágeno a nivel del hueso son la disminución de la matriz ósea, con alteración de la estructura ósea y mala mineralización (osteopenia), de forma que la resorción ósea predomina sobre la formación de hueso nuevo. Esto supone la presencia de la fragilidad ósea, fracturas frecuentes, deformidades óseas y talla baja. Las múltiples mutaciones descritas explican la gran heterogeneidad clínica de esta patología, existiendo desde formas mínimas a cuadros graves y letales. Otras consecuencias de la mutación de colágeno tipo I a distintos niveles son: hiperlaxitud de ligamentos y tendones, fragilidad vascular, disminución de la fuerza muscular, escleras azules y alteración de la dentinogénesis. Dependiendo de las características clínicas de las personas que presentan la enfermedad se han clasificado en varios grupos (Sillence, 1979).
OI – Tipo I OI – Tipo II OI – Tipo III OI – Tipo IV
Por lo general el tratamiento se centra en tres aspectos específicos: fisioterapia, tratamiento médico y tratamiento quirúrgico.

